
In część pierwsza tego artykułu, dokonałem przeglądu ram umownych i regulacyjnych stosowanych przez rząd USA przy początkowym opracowywaniu, produkcji i pozyskiwaniu zastrzyków mRNA Covid, wykorzystując umowy BioNTech/Pfizer do zilustrowania tego procesu.
Pokazałem, że zezwolenie na użycie awaryjne (EUA) zostało przyznane tym produktom na podstawie badań klinicznych i procesów produkcyjnych przeprowadzonych z nimi
- brak wiążących standardów prawnych,
- brak prawnie zakazanego nadzoru lub przepisów dotyczących bezpieczeństwa, oraz
- brak prawnego zadośćuczynienia ze strony producenta za potencjalne szkody.
W kolejnym artykule przedstawię szczegółową analizę podstawowej dokumentacji.
Inny organ transakcyjny/umowa (OTA): ścieżka przejęć wojskowych
Połączenia umowa pomiędzy rządem USA reprezentowanym przez Departament Obrony (DoD) a firmą Pfizer reprezentującą partnerstwo BioNTech/Pfizer w lipcu 2020 r. w sprawie zakupu „szczepionki zapobiegającej Covid-19” nie był zwykłą umową przejęcia.
Była to umowa w ramach Other Transaction Authority (OTA) – ścieżka przejęcia, która według Wytyczne Departamentu Obrony, jest używany od 1958 r. w celu „zezwolenia na wejście agencji federalnej”. transakcje inne niż umowy, dotacje lub umowy o współpracy".
[DODANO POgrubioną czcionkę]
Dokładny przegląd korzystania z OTA przez Departament Obrony, w tym jego ustawową historię, można znaleźć w: Raport Kongresowej Służby Badawczej z 22 lutego 2019 r. W tym raporcie, podobnie jak we wszystkich innych dyskusjach na temat OTA, określono, że jest to alternatywna ścieżka przejęć dla celów obronnych i wojskowych. Nie jest przeznaczony i nie był nigdy używany przed Covidem do niczego przeznaczonego głównie do użytku cywilnego.
Jeśli szukasz Przepisy OTA w Kodeksie USA, to jest ścieżka, którą pójdziesz:
Siły Zbrojne -> Ogólne prawo wojskowe -> Przejęcie -> Badania i inżynieria -> Umowy -> Upoważnienie Departamentu Obrony do realizacji określonych projektów prototypowych
Ta ścieżka prawna bardzo wyraźnie pokazuje, że przepisy OTA mają na celu pozyskiwanie prototypów badawczych i inżynieryjnych dla sił zbrojnych.
Według strony internetowej DARPA,
Departament Obrony ma uprawnienia do trzech różnych typów OT: (1) OT badawczych, (2) OT prototypowych i (3) OT produkcyjnych.
Te trzy typy OT reprezentują trzy etapy badań wstępnych, opracowania prototypu i ostatecznej produkcji.
W ramach tych trzech typów istnieją określone kategorie projektów, do których OTA może mieć zastosowanie:
- Pierwotnie wg Przegląd OTA dostarczony przez Departament Obrony, organ ds. innych transakcji „ograniczał się do stosowania do broni lub systemów uzbrojenia proponowanych do nabycia lub opracowania przez Departament Obrony”.
- OTA została później rozszerzona o „dowolny projekt prototypu bezpośrednio związany ze zwiększeniem efektywności misji personelu wojskowego oraz wspierających platform, systemów, komponentów lub materiałów proponowanych do nabycia lub opracowania przez Departament Obrony, lub z udoskonaleniem platform, systemów, komponentów lub materiałów używanych przez Siły Zbrojne.”
Jak dotąd nic z tego nie wygląda na ścieżkę nabycia milionów nowatorskich produktów medycznych przeznaczonych głównie do użytku cywilnego.
Czy istnieje wyjątek dotyczący cywilnego wykorzystania OTA, który może mieć zastosowanie do szczepionek mRNA na Covid?
Połączenia Ustawa o zezwoleniach na obronę narodową na rok budżetowy 2004 (P.L. 108-136) zawierał sekcję, w której uprawnienia do innych transakcji przyznano „szefowi agencji wykonawczej, która zajmuje się badaniami podstawowymi, badaniami stosowanymi, zaawansowanymi badaniami i projektami rozwojowymi”, które „mają potencjał ułatwienia obrony przed terroryzmem lub terroryzmem nuklearnym, biologicznym, nuklearnym, atak chemiczny lub radiologiczny.”
Przepis ten przedłużono do 2018 r., ale nie wydaje się, aby został przedłużony poza ten rok. Należy również pamiętać, że nawet w tym wyjątkowym przypadku użycia OTA bez DoD, sytuacja musi wiązać się z terroryzmem lub atakiem z użyciem broni masowego rażenia (CBRN).
Jakie inne przepisy dotyczące OTA mogą mieć zastosowanie?
Cytowany powyżej raport CRS z 2019 r. przedstawia ten wykres, pokazujący, że kilka agencji spoza DoD ma pewne OTA lub powiązane organy:
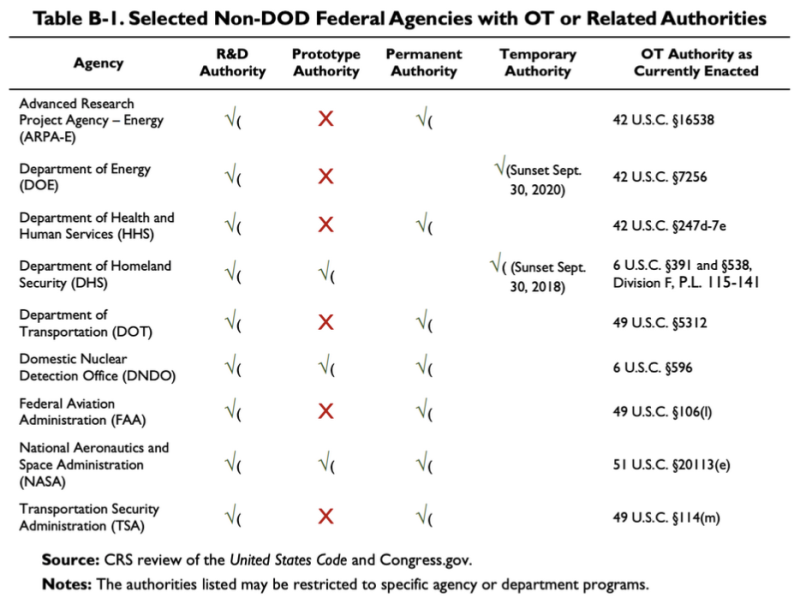
Zgodnie z tą tabelą Departament Zdrowia i Opieki Społecznej (HHS) prowadzi badania i rozwój (R&D), inne organy odpowiedzialne za transakcje. Prawo odnoszące się do Organem OT HHS jest 42 U.S.C. §247d-7e.
Gdzie znajduje się to prawo i co ono mówi?
Zdrowie publiczne i opieka społeczna -> Publiczna służba zdrowia -> Ogólne uprawnienia i obowiązki -> Współpraca z państwem federalnym -> Urząd ds. zaawansowanych badań i rozwoju biomedycznego (BARDA) -> Organy ds. transakcji
Jest zatem miejsce w prawie dotyczącym zdrowia i opieki społecznej ludności cywilnej, w którym OTA może mieć zastosowanie, chociaż jest ważne wyłącznie do badań i rozwoju, a nie do prototypów lub produkcji.
Prawo stanowi, że sekretarz BARDA ma uprawnienia OT
w odniesieniu do produktu, który jest lub może stać się kwalifikowanego środka zaradczego lub kwalifikowany produkt pandemiczny lub epidemiczny, działania, które głównie-
(i) są prowadzone po badaniach podstawowych i przedklinicznym rozwoju produktu; I
(ii) są związane z wytwarzaniem produktu na skalę handlową oraz w formie spełniającej wymogi regulacyjne na mocy ustawy federalnej Ustawa o żywności, lekach i kosmetykach [21 USC 301 i nast.] lub pod § 262 tego tytułu.
[DODANO POgrubioną czcionkę]
„Wymogi regulacyjne” wymienione w prawie oznaczają, że BARDA/HHS nie byłaby w stanie zawierać umów – nawet o charakterze badawczo-rozwojowym – na jakiekolwiek produkty medyczne (takie jak szczepionki mRNA), które nie przeszły rygorystycznych testów bezpieczeństwa i ścisłego nadzoru produkcyjnego.
„Partnerstwo” HHS z DoD obeszło przepisy dotyczące ochrony ludności cywilnej
Podsumowując trudną sytuację Innych Organów Transakcyjnych/Umów w odniesieniu do władz cywilnych ogólnie, a w szczególności szczepionek mRNA Covid:
- OTA został napisany i skodyfikowany jako sposób na zdobycie przez wojsko broni oraz innych niezbędnych systemów i sprzętu bez dużej biurokratycznej biurokracji. Obejmuje badania i rozwój, prototypy i późniejszą produkcję.
- Jedyna OTA dla agencji zdrowia publicznego jest przeznaczona dla HHS i obejmuje wyłącznie badania i rozwój, a nie prototypy czy produkcję.
- Nawet zezwolenie na badania i rozwój przyznane HHS w dalszym ciągu wymaga, aby produkty były wytwarzane „w formie spełniającej wymogi regulacyjne” dotyczące bezpieczeństwa leków i szczepionek.
Innymi słowy: nie ma możliwości, aby HHS mógł wykorzystać swoją bardzo ograniczoną platformę OTA do podpisania umów na setki milionów nowatorskich produktów medycznych.
Co więc zrobił HHS?
Jak zauważyło Biuro Odpowiedzialności Rządu (GAO) w swoim Raport z lipca 2021 r. na temat „Kontraktacji Covid-19:” HHS „nawiązał współpracę” z Departamentem Obrony, aby „wykorzystać władze OTA Departamentu Obrony… których brakowało HHS”. (str. 24)
Jakie są władze Departamentu Obrony w zakresie OT dotyczące produktów medycznych?
Jak już wspomniano, OTA ma pomóc wojsku w zdobyciu sprzętu i technologii bez wielu biurokratycznych kłopotów. Żadne z pierwotnych przepisów dotyczących OTA nie wspominało niczego innego niż „platformy, systemy, komponenty lub materiały” mające na celu „zwiększenie efektywności misji personelu wojskowego”.
Ale pięć lat przed Covidem wprowadzono wyjątkowe zastosowanie OTA:
W 2015, ogłosił DoD utworzenie konsorcjum przeciwdziałania medycznego CBRN, którego celem było wykorzystanie ścieżki przejęcia OTA do „współpracy z Departamentem Obrony w celu opracowania licencjonowanych przez FDA chemicznych, biologicznych, radiologicznych i nuklearnych środków zapobiegawczych”. [FDA = Agencja ds. Żywności i Leków]
Jak opisano w ogłoszeniu z 2015 r., obejmowało to „prototypowe technologie terapeutycznych medycznych środków zaradczych ukierunkowanych na cele wirusowe, bakteryjne i toksyny biologiczne będące przedmiotem zainteresowania Departamentu Obrony”. Lista czynników obejmowała najważniejsze patogeny broni biologicznej, takie jak wąglik, ebola i marburg.
W ogłoszeniu określono dalej, że „technologie prorozwojowe mogą obejmować zwierzęce modele chorób wirusowych, bakteryjnych lub biologicznych wywołanych toksynami oraz patogenezy (wiele dróg narażenia), testy, technologie diagnostyczne lub inne technologie platformowe, które można zastosować do opracowania zatwierdzonych lub licencjonowanych MCM [medyczne środki zaradcze]”.
Chociaż w niczym to nie przypomina produkcji 100 milionów nowych szczepionek do użytku cywilnego, zapewnia to OTA większą swobodę niż bardzo ograniczony organ ds. innych transakcji przyznany HHS.
Podczas gdy HHS OTA wymaga przestrzegania szeroko zakrojonych przepisów dotyczących rozwoju i produkcji, ścieżka OTA dla Departamentu Obrony w celu opracowania medycznych środków zaradczych wymaga jedynie „licencji FDA”.
Zatem, korzystając z innych organów transakcyjnych DoD, teoretycznie byłoby możliwe ominięcie wszelkich przepisów bezpieczeństwa – w zależności od wymagań dotyczących licencji FDA na produkt generowany przez OTA. Jak zobaczymy, w przypadku szczepionek mRNA Covid wydano zezwolenie na użycie w sytuacjach awaryjnych, które nie wymagało żadnego nadzoru prawnego dotyczącego bezpieczeństwa.
Zezwolenie na użycie awaryjne (EUA)
Oto jak Agencja ds. Żywności i Leków: (FDA) opisuje swoje uprawnienia EUA:
Artykuł 564 ustawy FD&C (21 USC 360bbb–3) umożliwia FDA wzmocnienie ochrony zdrowia publicznego przed czynnikami biologicznymi, chemicznymi, nuklearnymi i radiologicznymi.
Dzięki temu uprawnieniu EUA FDA może pomóc w zapewnieniu możliwości zastosowania medycznych środków zaradczych w sytuacjach awaryjnych w celu diagnozowania, leczenia lub zapobiegania poważnym lub zagrażającym życiu chorobom lub stanom spowodowanym przez czynniki biologiczne, chemiczne, nuklearne lub radiologiczne, gdy nie ma odpowiednich, zatwierdzonych oraz dostępne alternatywy (wśród innych kryteriów).
Niezwykle ważne jest, aby zrozumieć, że uprawnienia EUA zostały przyznane w 2004 r. w bardzo szczególnych okolicznościach związanych z gotowością na ataki bronią masowego rażenia, zwaną inaczej środkami CBRN (chemicznymi, biologicznymi, radiologicznymi i nuklearnymi).
Jak wyjaśniono w ustawie o zdrowiu Harvard Law,
Ostatecznie to wojna z terroryzmem doprowadziła do wydania zezwolenia na użycie w sytuacjach awaryjnych. Po wydarzeniach z 11 września 2001 r. i następujących po nich atakach pocztowych zawierających wąglika Kongres uchwalił Ustawa o projekcie Bioshield z 2004 r. Ustawa przewidywała przeznaczenie miliardów dolarów na zakup szczepionek w ramach przygotowań do ataku bioterrorystycznego oraz na gromadzenie zapasów środków zaradczych. Aby móc szybko działać w sytuacji awaryjnej, Kongres zezwolił FDA na dopuszczenie formalnie niezatwierdzonych produktów do użytku w sytuacjach awaryjnych ze względu na zagrożenie dla zdrowia i bezpieczeństwa publicznego (pod warunkiem ogłoszenia stanu nadzwyczajnego przez HHS). The rekord wskazuje, że Kongres skupił się konkretnie na zagrożeniu bioterroryzmem, a nie na przygotowaniach do naturalnie występującej pandemii.
Połączenia brzmienie ustawy EUA podkreśla fakt, że był on przeznaczony do użycia w sytuacjach związanych z bronią masowego rażenia. Oto 4 sytuacje, w których można wydać EUA:
- stwierdzenie przez Sekretarza Bezpieczeństwa Wewnętrznego, że ma miejsce sytuacja nadzwyczajna w kraju lub istnieje znaczne prawdopodobieństwo wystąpienia sytuacji awaryjnej w kraju, wiążącej się ze zwiększonym ryzykiem ataku z użyciem środka lub środków biologicznych, chemicznych, radiologicznych lub nuklearnych;
- stwierdzenie przez Sekretarza Obrony, że ma miejsce sytuacja nadzwyczajna wojskowa lub istnieje poważne ryzyko wystąpienia takiej sytuacji wojskowej, wiążącej się ze zwiększonym ryzykiem dla Stanów Zjednoczonych Zjednoczone sił zbrojnych, w tym personelu działającego na mocy tytułu 10 lub tytułu 50, ataku z:
- czynnik lub czynniki biologiczne, chemiczne, radiologiczne lub nuklearne; Lub
- agent lub agentów, którzy mogą powodować lub są w inny sposób powiązani z bezpośrednim zagrożeniem życia i szczególnym ryzykiem dla United Zjednoczone wojsko;
- ustalenie przez Sekretarz że istnieje zagrożenie zdrowia publicznego lub że istnieje poważne ryzyko wystąpienia stanu zagrożenia zdrowia publicznego, który ma wpływ lub może znacząco wpłynąć na bezpieczeństwo narodowe lub zdrowie i bezpieczeństwo Stanów Zjednoczonych Zjednoczone obywatele mieszkający za granicą, i które dotyczą czynnika lub czynników biologicznych, chemicznych, radiologicznych lub nuklearnych albo choroby lub stanu, który można przypisać takiemu czynnikowi lub czynnikom; Lub
- identyfikacji istotnego zagrożenia zgodnie z art. 319F–2 Ustawy Ustawa o publicznej służbie zdrowia [42 USC 247d–6b] wystarczający, aby wpłynąć na bezpieczeństwo narodowe lub zdrowie i bezpieczeństwo United Zjednoczone obywateli mieszkających za granicą.
Nigdzie w tych czterech sytuacjach nie ma wzmianki o naturalnie występującej epidemii, pandemii lub jakiejkolwiek innej sytuacji dotyczącej zdrowia publicznego, która nie jest spowodowana przez „czynnik(i) biologiczny, chemiczny, radiologiczny lub nuklearny”.
Czy SARS-CoV-2 może kwalifikować się jako taki czynnik?
Jeśli szukasz definicji „czynniki biologiczne”w amerykańskim kodeksie prawnym, będziesz podążać następującą ścieżką:
Przestępstwa i postępowanie karne -> Przestępstwa -> Broń biologiczna -> Definicje
Zatem w kontekście prawa Stanów Zjednoczonych termin „środki biologiczne” oznacza broń biologiczną, a użycie takich środków/broni jest uznawane za przestępstwo.
Wikipedia to podaje definicja:
Czynnik biologiczny (zwany także środkiem biologicznym, czynnikiem zagrożenia biologicznego, środkiem wojny biologicznej, bronią biologiczną lub bronią biologiczną) to bakteria, wirus, pierwotniaki, pasożyt, grzyblub toksyny, której można celowo użyć jako broni bioterroryzm or wojna biologiczna (BW).
Na jakiej podstawie prawnej wydano EUA dla szczepionek mRNA Covid?
Wydaje się, na podstawie przepisów dotyczących EUA, że żadna z czterech możliwych sytuacji opisanych w prawie nie może mieć zastosowania w przypadku produktu przeznaczonego do zapobiegania lub leczenia choroby wywołanej przez naturalnie występujący patogen.
Niemniej jednak na podstawie tego prawa zatwierdzono szczepionki mRNA Covid.
Biorąc pod uwagę cztery możliwości wymienione w prawie EUA, tą, która została zastosowana w przypadku „środków zaradczych” Covida była
C) ustalenie przez Sekretarz że istnieje zagrożenie zdrowia publicznego lub że istnieje poważne ryzyko wystąpienia stanu zagrożenia zdrowia publicznego, który ma wpływ lub może znacząco wpłynąć na bezpieczeństwo narodowe lub zdrowie i bezpieczeństwo Stanów Zjednoczonych Zjednoczone obywateli mieszkających za granicą i które wiąże się z czynnikiem lub czynnikami biologicznymi, chemicznymi, radiologicznymi lub nuklearnymi, bądź też chorobą lub stanem, który można przypisać takiemu czynnikowi lub czynnikom.
Kiedy zastosowane specjalnie w przypadku Covida, tak to zostało sformułowane:
Sekretarz Departamentu Zdrowia i Opieki Społecznej (HHS) stwierdził, że istnieje stan nadzwyczajny w zakresie zdrowia publicznego, który może znacząco wpłynąć na bezpieczeństwo narodowe lub zdrowie i bezpieczeństwo obywateli Stanów Zjednoczonych mieszkających za granicą i który jest związany z wirusem wywołującym koronaawirus Choroba 2019 (COVID-19)…
Nie ma tu wątpliwości, że „wirus powodujący COVID-19” jest uważany za odpowiednik „czynnika lub czynników biologicznych, chemicznych, radiologicznych lub nuklearnych”.
Należy również zauważyć, że „określenie stanu zagrożenia zdrowia publicznego” w ramach EUA jest całkowicie odrębne od jakichkolwiek innych deklaracji dotyczących stanu nadzwyczajnego zdrowia publicznego, takich jak te wydane przez WHO, rząd USA i w żaden sposób od nich nie zależy. i Prezydenta na początku pandemii Covid-19.
Zatem nawet jeśli WHO, rząd Stanów Zjednoczonych i Prezydent oświadczą, że pandemia się skończyła, nadal może zostać wydane zezwolenie na użycie w sytuacjach nadzwyczajnych, jeśli Sekretarz HHS w dalszym ciągu będzie twierdził, że istnieje sytuacja opisana w sekcji C).
Patrząc na wszystkie licencje EUA na setki produktów medycznych związanych z Covid-19bardzo trudno zrozumieć, w jaki sposób sekretarz HHS mógłby uzasadnić twierdzenie, że „istnieje stan nadzwyczajny w zakresie zdrowia publicznego, który może w znaczący sposób wpłynąć na bezpieczeństwo narodowe lub zdrowie i bezpieczeństwo obywateli USA mieszkających za granicą” w większości, jeśli nie we wszystkich, tych przypadków.
Dodatkowe „kryteria ustawowe” dotyczące udzielania przez FDA zezwolenia na użycie w sytuacjach awaryjnych
Gdy Sekretarz HHS oświadczy, że istnieje stan nadzwyczajny dotyczący zdrowia publicznego uzasadniający wydanie EUA, w oparciu o jedną z czterech sytuacji wymienionych w prawie, istnieją jeszcze cztery „kryteria ustawowe”, które muszą zostać spełnione, aby FDA wydała EUA . Oto jak FDA wyjaśnia te wymagania:
- Poważna lub zagrażająca życiu choroba lub stan
Aby FDA mogła wydać EUA, agent(y) CBRN, o którym mowa w deklaracji EUA Sekretarza HHS, musi być w stanie spowodować poważną lub zagrażającą życiu chorobę lub stan.
UWAGA: Kryterium to powtarza specyfikację środka CBRN, który prawnie definiuje się jako broń użytą do popełnienia przestępstwa.
- Dowód skuteczności
Produkty medyczne, które można uwzględnić w EUA, to te, które „mogą być skuteczne” w zapobieganiu, diagnozowaniu lub leczeniu poważnych lub zagrażających życiu chorób lub stanów, które mogą być spowodowane przez czynnik(-y) CBRN określony w oświadczeniu Sekretarza HHS awaria lub groźba sytuacji awaryjnej zgodnie z sekcją 564(b).
Standard „może być skuteczny” dla EUA zapewnia niższy poziom dowodów niż standard „skuteczności” stosowany przez FDA przy zatwierdzaniu produktów. FDA zamierza ocenić potencjalną skuteczność ewentualnego produktu EUA w każdym przypadku z osobna, stosując analizę ryzyka i korzyści, jak wyjaśniono poniżej.
[DODANO POgrubioną czcionkę]
PYTANIE PRAWNE: Jak ktokolwiek może zgodnie z prawem twierdzić, że produkt dopuszczony na mocy EUA jest „bezpieczny i skuteczny”, jeśli standard prawny dla EUA brzmi „może być skuteczny”, a FDA deklaruje, że jest to „niższy poziom dowodów” niż stosowany standard do regularnych zatwierdzeń produktów?
- Analiza ryzyka i korzyści
Produkt może zostać uznany za EUA, jeśli Komisarz ustali, że znane i potencjalne korzyści produktu stosowanego do diagnozowania, zapobiegania lub leczenia zidentyfikowanej choroby lub stanu przewyższają znane i potencjalne ryzyko związane z produktem.
Przy ustalaniu, czy znane i potencjalne korzyści produktu przewyższają znane i potencjalne ryzyko, FDA zamierza patrzeć na podstawie całości dowodów naukowych, aby dokonać ogólnego określenia stosunku ryzyka do korzyści. Taki dowód, który mogłoby powstać z różnych źródeł, może zawierać (m.in.): wyniki badań klinicznych krajowych i zagranicznych, dane dotyczące skuteczności in vivo na modelach zwierzęcych oraz dane in vitro, dostępne do rozpatrzenia przez FDA. FDA oceni również jakość i ilość dostępne dowody, biorąc pod uwagę obecny stan wiedzy naukowej.
[DODANO POgrubioną czcionkę]
UWAGA PRAWNA: Nie ma normy prawnej ani definicji prawnych określających, co oznacza, że „znane i potencjalne korzyści” przewyższają „znane i potencjalne ryzyko”. Nie ma również jakościowej ani ilościowej definicji prawnej tego, co stanowi akceptowalny „dostępny dowód”, na którym „może” opierać się analiza ryzyka i korzyści. Rzeczywistych dowodów może nie być żadnych, ale istnieje przekonanie, że produkt niesie ze sobą wiele potencjalnych korzyści, a nie wiele potencjalnego ryzyka, co spełniałoby ten „wymóg ustawowy”.
- Żadnych alternatyw
Aby FDA mogła wydać EUA, nie może istnieć odpowiednia, zatwierdzona i dostępna alternatywa dla produktu kandydującego do diagnozowania, zapobiegania lub leczenia choroby lub stanu. Potencjalny produkt alternatywny można uznać za „niedostępny”, jeżeli zapasy zatwierdzonego produktu alternatywnego są niewystarczające, aby w pełni zaspokoić zapotrzebowanie w sytuacjach nadzwyczajnych.
PYTANIE PRAWNE: Oprócz rażącego i potencjalnie przestępczego oczerniania/zakazu alternatywnych metod leczenia Covid-19, takich jak iwermektyna i hydroksychlorochina, w którym momencie istniała zatwierdzona alternatywa „zapobiegania Covid-19” (jedyne do czego zakupiono szczepionki mRNA ) – na przykład Paxlovid – co spowodowałoby, że EUA dla szczepionek mRNA przestałoby być legalne?
Oto jak wszystkie te „ustawowe kryteria” zostały spełnione w rzeczywistości Zezwolenie na stosowanie w sytuacjach awaryjnych szczepionek mRNA BioNTEch/Pfizer Covid:
Doszedłem do wniosku, że awaryjne zastosowanie szczepionki Pfizer-BioNTech przeciwko Covid-19 w celu zapobiegania chorobie Covid-19, podawanej zgodnie z opisem w Zakresie zezwolenia (część II), spełnia kryteria wydania zezwolenia zgodnie z sekcją 564 lit. c) ustawy ustawy, ponieważ:
- SARS-CoV-2 może powodować poważną lub zagrażającą życiu chorobę lub stan, w tym ciężką chorobę układu oddechowego, u ludzi zakażonych tym wirusem;
- Na podstawie całości dowodów naukowych dostępnych FDA można zasadnie sądzić, że szczepionka Pfizer-BioNTech na Covid-19 mogą być skuteczne w zapobieganiu Covid-19oraz że w przypadku stosowania zgodnie z warunkami opisanymi w niniejszym zezwoleniu znane i potencjalne korzyści szczepionki Pfizer-BioNTech na Covid-19 w przypadku stosowania w celu zapobiegania COVID-19 przewyższają znane i potencjalne ryzyko; I
- Nie ma odpowiedniej, zatwierdzonej i dostępnej alternatywy dla awaryjnego stosowania szczepionki Pfizer-BioNTech przeciwko Covid-19 aby zapobiec Covid-19.
[DODANO POgrubioną czcionkę]
UWAGA: Jedynym kontekstem, w którym FDA rozważała potencjalne korzyści i ryzyko związane ze szczepionką i w którym stwierdziła, że „może być skuteczna”, był w zapobieganiu Covid-19.
Nie wzięto pod uwagę, nie ma dowodów na rzeczywiste lub potencjalne korzyści ani nie ustalono, że szczepionka ma jakąkolwiek potencjalną skuteczność w jakimkolwiek innym działaniu, w tym: zmniejszaniu ryzyka ciężkiej choroby, zmniejszaniu ryzyka hospitalizacji, zmniejszaniu ryzyka śmierci , zmniejszając ryzyko wystąpienia jakichkolwiek schorzeń faktycznie lub potencjalnie związanych z Covid-19.
DLATEGO można rozsądnie kwestionować legalność jakichkolwiek twierdzeń, że szczepionka jest „bezpieczna i skuteczna” w kontekście innym niż „kiedy jest stosowana w celu zapobiegania COVID-19” – czego szczepionki NIE ROBIŁY już wkrótce po ich wprowadzeniu wprowadzony.
Czy gdyby ludziom powiedziano, że szczepionki mRNA BioNTech/Pfizer są „bezpieczne i skuteczne” w czymkolwiek innym niż zapobieganie Covid-19 i gdyby grożono im jakimikolwiek konsekwencjami za niezastosowanie szczepionki w innym celu niż zapobieganie Covid-19, czy mają uzasadniony argument, że zostali bezprawnie zmuszeni do nabycia niezatwierdzonego produktu w oparciu o fałszywe twierdzenia?
Wymagania trzeciego poziomu dotyczące uprawnień EUA dla niezatwierdzonych produktów
Kiedy już otrzymamy deklarację nadzwyczajną specyficzną dla EUA i kiedy FDA zadeklaruje, że produkt może być skuteczny i że wszelkie dostępne dowody (od zera do nieskończoności) wykażą, że korzyści z niego płynące przewyższają ryzyko (określone na podstawie tego, co zdaniem FDA może to spowodować) be), istnieje jeszcze jeden poziom regulacji niezwiązanych z bezpieczeństwem i skutecznością.
Oto jak Raport Congressional Research Service z 2018 r. na temat EUA wyjaśnia to:
FFDCA §564 nakazuje FDA nałożenie pewnych wymaganych warunków w EUA i dopuszcza dodatkowe, uznaniowe warunki, w stosownych przypadkach. Wymagane warunki różnią się w zależności od tego, czy licencja EUA dotyczy niezatwierdzonego produktu, czy niezatwierdzonego użycia zatwierdzonego produktu. W przypadku niezatwierdzonego produktu warunki stosowania muszą:
(1) zapewnić, że pracownicy służby zdrowia podający produkt otrzymają wymagane informacje;
(2) zapewnić, że osoby, którym podaje się produkt, otrzymały wymagane informacje;
(3) zapewnić monitorowanie i zgłaszanie zdarzeń niepożądanych związanych z produktem; I
(4) zapewniać prowadzenie rejestrów i raportowanie przez producenta.
PYTANIE PRAWNE: Czym dokładnie są „wymagane informacje”? Wiemy, że ludzie zostali poinformowani, że szczepionki otrzymały zezwolenie na stosowanie w sytuacjach nadzwyczajnych. Ale czy powiedziano im, że oznacza to „niższy poziom dowodów” niż jest to wymagane w przypadku „bezpiecznych i skutecznych” oświadczeń dotyczących innych produktów medycznych? Czy zostali poinformowani, że istnieją różne poziomy „bezpiecznego i skutecznego” w zależności od tego, czy produkt posiada zezwolenie EUA, czy inny rodzaj autoryzacji?
UWAGA: Prawo wymaga, aby istniał sposób monitorowania i zgłaszania zdarzeń niepożądanych. Nie określa jednak, kto monitoruje, jakie są standardy raportowania i jaki jest próg podejmowania działań na podstawie raportów.
EUA w porównaniu do każdej innej ścieżki zatwierdzania leków/szczepionek
Jako badacz/pisarz Sasza Łatypowa zauważył, wiele osób było zdezorientowanych pojęciem EUA, ponieważ brzmi ono bardzo podobnie do EAU, co oznacza „Expanded Access Use”. Jest to rodzaj zezwolenia wydawanego produktom medycznym w przypadku pilnej potrzeby określonej grupy pacjentów (np. pacjentów z nowotworem w stadium IV, których oczekiwana długość życia jest mierzona w miesiącach), którzy są skłonni zaryzykować wystąpienie działań niepożądanych, a nawet śmierć w zamian za dostęp. na eksperymentalne leczenie.
Autoryzacja do korzystania w sytuacjach awaryjnych nie jest w żaden sposób powiązana z korzystaniem z rozszerzonego dostępu ani do niego nie przypomina.
Różne ścieżki prawne dopuszczania produktów medycznych są starannie przedstawione w tabeli wyróżnionej przez badacza prawa Katarzyna Watt. Tabela jest częścią prezentacji na rok 2020 podczas wspólnej sesji edukacyjnej FDA-CDC: Aktualizacje przepisów dotyczące stosowania medycznych środków zaradczych.
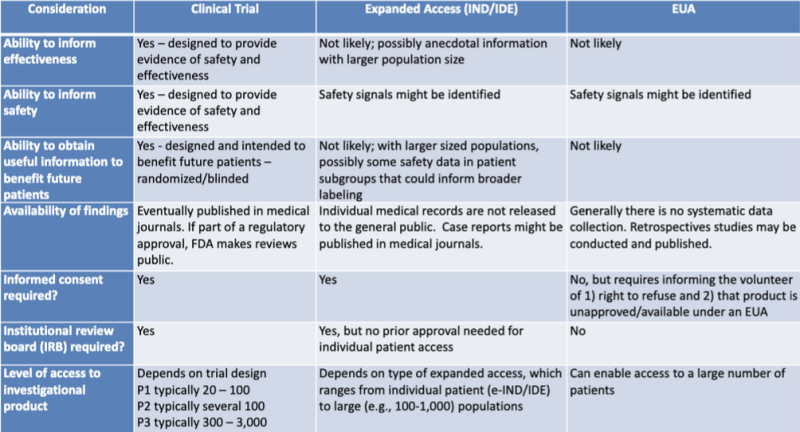
Tabela ta pokazuje bardzo wyraźnie, że proces EUA prawdopodobnie nie dostarczy informacji dotyczących skuteczności produktu, nie ma na celu dostarczenia dowodów bezpieczeństwa, prawdopodobnie nie dostarczy przydatnych informacji z korzyścią dla przyszłych pacjentów, nie wymaga systematycznego gromadzenia danych, nie wymaga badań retrospektywnych, bez świadomej zgody i bez instytucjonalnej komisji odwoławczej.
Co więcej, w 2009 Instytut Medyczny publikacji Narodowej Akademickiej, również podkreślony przez Watta, zatytułowany „Medyczne środki zaradcze: wydawanie zezwolenia na użycie w sytuacjach awaryjnych i model pocztowy – podsumowanie warsztatów”, znajdujemy to stwierdzenie na s. 28:
Należy pamiętać, że licencja EUA nie jest częścią ścieżki rozwoju; jest to całkowicie odrębna jednostka, używana wyłącznie w sytuacjach awaryjnych i nie będąca częścią procesu zatwierdzania leku.
Czy to oznacza, że zatwierdzenie środków zaradczych na Covid-19 opartych na EUA było nielegalne? Czy to oznacza, że nie ma prawnego sposobu, aby twierdzić, że produkt EUA jest „bezpieczny i skuteczny”, ponieważ NIE JEST CZĘŚCIĄ PROCESU ZATWIERDZANIA LEKU?
Wnioski
Jest to całkowicie oczywiste, biorąc pod uwagę wszystkie informacje zawarte w tym artykule i w poprzednim Część 1, że szczepionki mRNA BioNTach/Pfizer Covid zostały opracowane, wyprodukowane i dopuszczone na mocy prawa wojskowego zastrzeżonego dla sytuacji nadzwyczajnych związanych z wojną biologiczną/terroryzmem, a nie chorobami występującymi naturalnie, dotykającymi całą populację cywilną.
Dlatego przestrzeganie przepisów i nadzór, których spodziewamy się, gdy produkt zostanie uznany za „bezpieczny i skuteczny” dla całej populacji cywilnej, nie były prawnie wymagane.
Czy tę analizę można wykorzystać do zakwestionowania legalności „bezpiecznego i skutecznego” twierdzenia tych urzędników rządowych, którzy wiedzieli, co oznacza EUA? Czy istnieją inne konsekwencje prawne?
Mam nadzieję.
Co ważne, w dotychczas wniesionych skargach prawnych dotyczących szczepionek mRNA na Covid nie wydano (o ile mi wiadomo) żadnych orzeczeń określających, czy prawo wojskowe, takie jak OTA i EUA, może mieć zastosowanie w sytuacjach cywilnych. Jednakże pojawiło się oświadczenie sędziego Sądu Rejonowego Michaela Truncale w jego wyroku umorzenie sprawy sygnalisty Brook Jackson przeciwko Ventavii i Pfizer, warto o tym pamiętać.
W tym miejscu sędzia przyznaje, że umowa dotycząca szczepionek mRNA BioNTech/Pfizer była wojskową OTA, ale odmawia wydania orzeczenia w sprawie jej zastosowania w okolicznościach pozamilitarnych (choroba występująca naturalnie, 100 milionów dawek, w większości nieprzeznaczonych do użytku wojskowego), na mocy których zostało wydane:
Fakt, że szczepionkę otrzymał zarówno personel wojskowy, jak i cywile, nie oznacza, że zdobycie szczepionki nie miało znaczenia dla zwiększenia efektywności misji wojskowej. Co ważniejsze, pani Jackson faktycznie zwraca się do Trybunału o uchylenie decyzji Departamentu Obrony o skorzystaniu z uprawnienia do przeprowadzania innych transakcji w celu zakupu szczepionki firmy Pfizer. Jak jednak od dawna podkreślał Sąd Najwyższy Stanów Zjednoczonych, „złożone, subtelne i profesjonalne decyzje dotyczące składu, wyszkolenia, wyposażenia i kontroli sił zbrojnych są w istocie profesjonalnymi osądami wojskowymi”. Gilligan przeciwko Morganowi, 413 U.S. 1, 10 (1973). Tym samym „trudno wyobrazić sobie obszar działalności państwa, w którym sądy mają mniejsze kompetencje”. ID. Trybunał nie zawetuje orzeczeń Departamentu Obrony dotyczących skuteczności misji podczas stanu nadzwyczajnego w kraju.
To tylko jedna z wielu przeszkód prawnych, które pozostają w walce o ostateczne zakazanie wszystkich produktów mRNA zatwierdzonych podczas kryzysu związanego z Covid-19 oraz wszelkich kolejnych produktów mRNA, których zatwierdzenie opierało się na procesie zatwierdzania Covid-19.
Opublikowane pod a Creative Commons Uznanie autorstwa 4.0 Licencja międzynarodowa
W przypadku przedruków ustaw link kanoniczny z powrotem na oryginał Instytut Brownstone Artykuł i autor.








