

W ciągu ostatnich dziesięcioleci mojej kariery spędziłem niezliczone godziny, pracując na rzecz ochrony Amerykanów, badając bezpieczeństwo narkotyków. Moja edukacja i kariera prowadziły mnie przez około pół tuzina uniwersytetów, Big Pharma i FDA pod trzema administracjami prezydenckimi. Bezpieczeństwo leków uwzględnia, dlaczego jedna osoba może zażywać produkt farmaceutyczny i nie mieć żadnych działań niepożądanych, podczas gdy inna osoba może przyjmować ten sam produkt, ale mogą wystąpić u niego działania niepożądane, aż do trwałego kalectwa lub śmierci włącznie. Domyślnie badanie bezpieczeństwa leków uwzględnia także niekliniczne aspekty wytwarzania i jakości leków.
Ponieważ jakość leku jest istotnym czynnikiem w ocenie bezpieczeństwa leków, moje działania mające na celu ochronę Amerykanów doprowadziły do opracowania konceptualizacji i założenia pierwszego na świecie „apteka analityczna” którego misją jest naukowa weryfikacja produktów farmaceutycznych z takich miejsc jak Indie i Chiny przed wydaniem ich pacjentom. Niestety, przedkładanie wyższości nad etykę i ochronę pacjentów doprowadziło do błędów w zarządzaniu finansami tej firmy rozległy Naruszenia FDA i bycie oskarżanym przez sędziów o robienie fałszywe twierdzenia naukowe (z których wszystko nieprzypadkowo wydarzyło się po moim wyjściu).
Bez zewnętrznego potwierdzenia jakości leku Amerykanie są całkowicie zależni od FDA i producentów w zakresie oceny i potwierdzania czystości produktu. Wykazano, że bezpieczeństwo leków jest godnym uwagi problemem, jeśli chodzi o zastrzyki mRNA Covida. Niestety, jeśli ktoś chciał przeprowadzić własną analizę zastrzyków mRNA, to nie nie mają odpowiednio szczegółowej listy składników, z którą można by je porównać, ani nawet dostępu do ustalonej metodologii regulacyjnej dotyczącej prawidłowego testowania czystości.
To dlatego, że producenci i FDA bierze pod uwagę wszystkie składniki tych zastrzyków mRNA, w tym sekwencję mRNA i właściwości nanocząstek lipidowych (LNP), w tym okres półtrwania, struktury LNP, modyfikacje powierzchni, liczbę/typ(y) LNP na dawkę i punkty mocowania na nić mRNA, nieokreśloną lub „tajemnicą handlową”.
Ponadto FDA bierze również pod uwagę metodologie tajemnica handlowa dotycząca sposobu testowania zastrzyków mRNA pod kątem czystości.
Wsparcie ponadpartyjne i setki miliardów dolarów podatników, ale BRAK przejrzystości?
Tajemnica dotycząca mRNA Covida istnieje, mimo że administracja Trumpa i Bidena zaproponowała pełną przejrzystość w zakresie zastrzyków mRNA do tego stopnia, że zniesie prawa własności intelektualnej mRNA Covida. Mimo to zarówno FDA, jak i producenci zezwalają/utrzymują patenty, w tym podstawowe dane na temat tych zastrzyków, jako tajemnicę handlową. Robią to pomimo otrzymania wszystkich producentów szczepionek na Covid setki milionów dolarów podatników według Forbesa/statysty publikacje.
Studiowanie epidemiologii bezpieczeństwa leków jest wystarczająco trudne. Bez sprawdzalnej czystości/spójności produktu pełna ocena bezpieczeństwa jest niemożliwa.
Pełna przejrzystość wszystkich składników i środków kontroli jakości jest ważna nie tylko dlatego, że były one w dużej mierze finansowane przez podatników w wysokości setek milionów dolarów, ale także dlatego, że pojawiło się mnóstwo pytań dotyczących bezpieczeństwa i skuteczności zastrzyków mRNA Covida.
Oprócz tego, że były wyjątkowo złożone, ich zatwierdzenie zostało później przyspieszone przez organy regulacyjne mniej niż rok. Większość leków i szczepionek zazwyczaj jest powszechnie dostępna dziesięć lat w celu pełnego przetestowania bezpieczeństwa/skuteczności oraz przeglądu i zatwierdzenia. Oprócz tego, że składniki są całkowicie nowatorskie, bardzo złożone i pierwsze tego typu do podawania na masową skalę, opracowanie m.in. długoterminowe oceny bezpieczeństwa klinicznego/toksyczności oraz przeglądy epidemiologiczne zostały przyspieszone i prawdopodobnie nie zostały w pełni wyjaśnione przed wypuszczeniem na rynek.
Weryfikacja składników, przejrzystość i „prawdziwość” FDA mają precedensy sięgające XIX wieku:
Weryfikacja analityczna i przejrzystość składników, czyli „prawda w oznakowaniu”, gdzie znajduje się zawartość butelki wymagany pasować do wymienionych składników sięga czasów poprzedzających utworzenie FDA, czyli do roku 1862. Dzisiejsza FDA tak naprawdę narodziła się z tego, co zaczęło się jako pojedynczy pracownik „Departamentu Chemii” zatrudniony w Departamencie Rolnictwa Stanów Zjednoczonych.
Fałszerstwo, (zmienione lub toksyczne składniki) błędna marka (zawiera fałszywą etykietę lub w inny sposób wprowadza w błąd lub zawiera nieprawidłowe oświadczenia medyczne) lub błędne oznakowanie (zawiera składniki, które nie są wymienione na etykiecie produktu) mają długą i brzydką historię w Ameryce. Uważano, że agresywność osiągnęła swój szczyt na początku lub w połowie XIX wieku – a przynajmniej wtedy można ją było zidentyfikować – ponieważ dopiero w 19 roku opracowano procesy techniczne umożliwiające analizę i wykrywanie oszustw związanych ze składnikami. Wcześniej tak zwani „wędrujący szamani” nazywający siebie „lekarzami” (zawsze z wątpliwymi lub nieistniejącymi referencjami) sprzedawali butelki z produktami „lekami na wszystko”, których etykiety składników podawały jedynie mglistą lub nieszkodliwą zawartość, taką jak „witaminy""ekstrakty ziołowe,"Lub"olej wężowy” – lub często nie mają w ogóle listy składników.
W tamtych czasach wielu pobożnych, purytańskich mieszkańców Nowej Anglii, którzy z powodów religijnych tak robili nigdy dotkną alkoholu, kupiliby te rozwiązania od handlarzy handlarzami i nieświadomie daliby się nabrać na spożycie roztworów zawierających nie tylko alkohol, ale także narkotyki, takie jak opium i/lub kokaina. Pod pozorem poprawy absurdalnie szerokiej gamy dolegliwości, zamiast tego u pacjentów rozwinęło się karzące uzależnienie i/lub w inny sposób pierwsi „handlarze narkotyków” wpłynęli negatywnie na ich zdrowie.
W miarę narastania problemu rząd federalny zaczął zwracać na to uwagę. Ostatecznie, Ustawa o czystej żywności i narkotykach została uchwalona w 1906 r. i doprowadziła do utworzenia Agencji ds. Żywności i Leków (FDA).
[FDA miała szkolenie obowiązek zapewnienia, że leki są opatrzone zgodnymi z prawdą oświadczeniami na etykietach i spełniają określone standardy czystości i mocy.
Pamiętajcie, że prawie 120-latek wymóg dotyczący zgodnego z prawdą etykietowania oraz część „czystości” ustawy o czystej żywności i lekach z 1906 r., gdy czytasz dalej o testach weryfikacyjnych mRNA i przejrzystości składników.]
Jakie „prawdziwe” i „czyste” testy weryfikacyjne składników są przeprowadzane w przypadku produktów regulowanych przez FDA?
W 2021 r. FDA zdecydowała się rozpocząć monitorowanie jakości farmaceutycznej w Ameryce za pośrednictwem: zbiór zdalny of przesłanie próbek pocztą na leki w zastępstwie inspekcji obiektów na żywo w związku z pandemią Covid. Czy to było legalne? Czy można to kiedykolwiek uznać za naukowo właściwe? Dziś, pomimo wygaśnięcia pandemii, prowadzone są obecnie jedyne oficjalne badania uwalniania farmaceutycznego każdy Farmaceutyczny mRNA Covida pojawia się do nadal może zostać wykonane przez FDA za pośrednictwem dostawcy dostarczonego przez producenta, „wysłane” próbka wg A zrzut ekranu aktualnej strony internetowej FDA. Oczywiście metoda pobierania próbek „wysyłana pocztą” jest znacznie inna i potencjalnie mniej niezawodna niż bezpośrednie pobieranie próbek metodą bezpośrednią, osobistą. Pomimo tego FDA twierdzi, że „najwyższy na całym świecie standard pobierania próbek i testowania".
Co więcej, FDA proponuje dalsze udoskonalanie swojej polityki zdalnego testowania „wysyłanego pocztą” za pomocą: nowo zaproponowany dokument zawierający wytyczne.
Chociaż istnieje on jedynie jako „projekt” dokumentu FDA, oficjalne strony internetowe FDA to pokazują wydaje się, że wysyłka próbek jest już realizowana co najmniej od stycznia 2021 r. Wydaje się, że FDA uznaje wyniki przesłanych pocztą testów za niezależną weryfikację.
Ponadto na dole pierwszej strony projektu FDA dokument proponuje rozszerzenie „testowania zdalnego”. Obecnie wymienia każdy Dział FDA ds. regulacji produktów w FDA, co sugeruje, że jest to propozycja polityczna obejmująca całą agencję.
Pełna lista obejmuje:
- Biuro Spraw Regulacyjnych
- Biuro Polityki Żywnościowej i Reagowania
- Biuro Produktów Kombinowanych
- Centrum Oceny i Badań Biologicznych
- Centrum oceny leków i badań
- Centrum Urządzeń i Zdrowia Radiologicznego
- Centrum Bezpieczeństwa Żywności i Żywienia Stosowanego
- Centrum Wyrobów Tytoniowych
- Centrum Medycyny Weterynaryjnej
Czy pobieranie próbek do kontroli jakości „wysyłanych pocztą” przez FDA jest właściwe? Co by było, gdyby inspekcje restauracji Departamentu Zdrowia Stanów odzwierciedlały politykę FDA?
Ta metodologia pobierania próbek „pocztą” jest podobnie absurdalna, na przykład w przypadku stanowego departamentu zdrowia monitorującego restauracje, prosząc je o okresowe „wysyłanie” różnych pozycji z menu do placówki przeprowadzającej testy, aby departamenty zdrowia mogły przetestować potencjalną żywność powstałe skażenie i/lub proszenie restauracji o obietnicę samodzielnego przetestowania pozycji menu. A co by było, gdyby ta restauracja była w Chinach? A co by było, gdyby ta restauracja znajdowała się w Indiach? Lub jakikolwiek inny kraj, o którym wiadomo, że ma fatalna historia oszustw i kontroli jakości problemy?
Taka metodologia byłaby nie do przyjęcia zarówno dla restauracji, jak i firm farmaceutycznych, z powodów między innymi oczywistych: producenci mogliby przesyłać preferowane przez siebie próbki – niekoniecznie reprezentatywne próbki partii. To oczywiście nie to samo, co inspektorzy FDA pobierający próbki podczas niezapowiedzianych inspekcji całego obiektu.
Zgodnie z analogią do restauracji, oczywiście wszystkie restauracje tak zrobią przedłożyć próbki klasy „A”. które niekoniecznie byłyby reprezentatywne dla tego, co otrzymują konsumenci.

Kontrola jakości: Co to jest „testowanie wydania” farmaceutycznego i dlaczego jest ważne?
Obecnie FDA nadzoruje jakość i zawartość $2.7 trylion wartość produktu rocznie, ale wydaje się, że pomija krytyczne oceny i wyniki weryfikacji składników. FDA ma chronić Amerykanów poprzez prowadzenie wszechstronny testy analityczne jako suma kontrolna zapewniająca dokładność składników. Wyniki tego powinny być przejrzyste dla podatników finansujących 6.6 miliarda dolarów FDA budżet. Ta weryfikacja naukowa nazywana jest „farmaceutyczną”testowanie wydania.” Testowanie wersji to termin techniczny odnoszący się do procesu obejmującego różnorodne analizy instrumentalne kompleksowo testować produkty pod kątem czystości, stężenia, konsystencji, tożsamości i wszelkiego rodzaju zanieczyszczeń.
Cała FDA zrodziła się z jednego pracownika „Wydziału Chemii” z 1862 roku oraz potrzeby przejrzystości i weryfikacji składników. Dziś ten pracownik rozrósł się do rangi: cały dział FDA składający się z 1,300 naukowców i personelu pomocniczego przypuszczalnie poświęcony weryfikacji składników poprzez badanie uwalniania farmaceutycznego. FDA Biuro Jakości Farmaceutycznej (OPQ) ma zapewnić, że farmaceutyki dokładnie odpowiadają zawartości wymienionych składników, bez zmienności jakości/zanieczyszczenia (jakościowej) lub zawartości (jakościowej). Wymagające tego przepisy są bardzo szczegółowe i szczegółowe 21 CFR § 201.10.
Jak FDA weryfikuje zastrzyki mRNA w celu kontroli jakości:
Wyniki kontroli jakości testów iniekcji mRNA były szczególnie krytyczne, ponieważ są duże, złożone i wykonano je szybko. Podczas gdy podatnicy polegają na FDA w zakresie weryfikacji jakości wstrzyknięcia mRNA i udostępniania wyników, FDA wydaje zobowiązany do ochrony składników producentów kosztem nawet najbardziej podstawowej przejrzystości dotyczącej produktów mRNA Covid. Chociaż wydaje się, że FDA zbiera próbki, ich metodologia „wysyłania pocztą” jest zasadniczo błędna. Ponadto FDA nie udostępnia wyników tych testów nigdzie, gdzie mógłbym je zlokalizować.
Innymi słowy: podczas pandemii, kiedy zupełnie nowe, szeroko stosowane zastrzyki mRNA były wysyłane do Amerykanów z „prędkością osnowy” i kiedy Ameryka w największym stopniu polegała na obowiązkach FDA w zakresie jakości/regulacji, FDA akceptowała przesłane przez siebie „wysłane pocztą” w” testach i/lub wynikach kontroli jakości. Czy FDA tego nie wzięła pod uwagę? Producenci mRNA przyznali, że „trudno im reagować na produkcję i „starali się”, aby dotrzymać kroku z procesami produkcyjnymi? Producenci składników mRNA stwierdzili ponadto, że wysiłki mające na celu zaspokojenie potrzeb były „bezprecedensowe”.
Tego typu stwierdzenia nie dają zaufania konsumentów do jakości i ilustrują ogromne zwiększenie skali tych złożonych produktów, co powinno uzasadniać szczególnie czujny oraz osobista kontrola FDA obiektów i wytwarzanych produktów, niezależnie od pandemii czy nie. Na przykład jeden z producentów składników mRNA stwierdził, że nagle zwiększył produkcję 50 krotnie.
Czy w środku tej nowatorskiej technologii wprowadzanej w „prędkości osnowy” żaden z 1,300 naukowców OPQ z FDA nie domagał się kontroli na żywo lub przynajmniej nie oferował niczego innego niż proszenie o potencjalnie wątpliwe próbki „wysłane pocztą” dla testów?
Oczywiste pytanie brzmi: dlaczego FDA nie pobrała próbek bezpośrednio? Nawet w obliczu pandemii FDA mogła przeprowadzić inspekcję obiektów w kombinezonach ochronnych lub – lub na miejscu początku. najmniej – zdecydowało się na pobieranie próbek z aptek, szpitali lub magazynów dystrybutorów.
Ukryta metodologia testowania składników do wstrzykiwań mRNA:
Poza brakiem wyników badań i wątpliwymi wynikami pobierania próbek przesłanymi pocztą – FDA tak dodatkowo ukrywanie swojej zatwierdzonej metodologii, uniemożliwiając innym przeprowadzanie własnych, niezależnych analiz dotyczących jakości/czystości zastrzyków mRNA.
Niezależne analizowanie leków pod kątem czystości i potencjalnego zanieczyszczenia w porównaniu z listą składników, to coś, co próbowałem zrobić sam, kiedy opracowywałem pierwszą na świecie apteka analityczna. Ponieważ jednak zastrzyki mRNA to nowatorska technologia z niezbyt przejrzystą listą składników, metodologia testowania, którą należy zastosować, nie jest prosta, jak miałoby to miejsce w przypadku innych leków małocząsteczkowych. Każdy, kto próbuje sprawdzić przechowywanie, stabilność, specyficzność, chemię, czułość lub nawet podstawową metodologię walidacji testów i/lub wyników, jest blokowany przez raport FDA zawierający absurdalnie inwazyjne redakcje, co sprawia, że nawet najbardziej podstawowe naukowe zrozumienie potencjalnej oceny wyników lub przeprowadzenia badania niemożliwe.
Przejmującym przykładem wizualnym jest pojedyncza zredagowana strona w dłuższym podsumowaniu regulacyjnym FDA (pokazanym poniżej) Dokument 127 stron (z których udostępniono tylko 63 strony, z których około 63% zostało zredagowanych) jak ocenić czystość, stężenie i inne miary analityczne zastrzyków mRNA.
Ci, Redakcja FDA (b) (4). określone szczegółowe redakcje użyte do „chronić tajemnice handlowe i poufne informacje handlowe lub finansowe.” Ale czy naprawdę właściwe jest nazywanie tego „komercyjnym”, jeśli badania/rozwój/produkt zostały sfinansowane? setki milionów dolarów podatników?

Bez listy składników lub metodologii testowania nikt inny spoza FDA i producentów nie może dokładnie wiedzieć, jak sprawdzić produkt fałszerstwo (zmienione lub toksyczne składniki) lub błędne oznakowanie (ponieważ pełna lista składników, w tym sekwencja nukleotydów i konfiguracje nanocząstek lipidowych są szczególnie niejasne na etykiecie produktu).
Brak metodologii jest szczególnie kłopotliwy, ponieważ wykazały to nowe, wstępne dane wykorzystujące niezależną metodologię Zanieczyszczenie DNA w zastrzykach mRNA Covid.
Tak więc, jeśli osoba z zewnątrz twierdziła, że przeprowadziła testy i znalazła zanieczyszczenia w zastrzykach mRNA i poprosiła FDA lub producentów o odpowiedź, spotkałaby się z odpowiedzią zawierającą coś w rodzaju:
- Do wyciągnięcia wniosków nie wykorzystałeś sprawdzonej/odpowiedniej metodologii testów, dlatego Twoje analizy są nieważne.
W tym celu niezależne laboratorium będzie próbowało uzyskać metodologię badań z dokumentacji zatwierdzonej przez FDA (tj. pełnego dokumentu zawierającego Postać 5) pytając: „OK, chciałbym to przetestować, stosując zatwierdzoną przez Ciebie metodologię; powiesz nam, co to jest?”
- FDA lub producent odpowiedziałby mniej więcej w następujący sposób: „To, co jesteśmy skłonni ujawnić na temat zastosowanej metodologii, a które nie jest poufne, można znaleźć w Internecie lub za pośrednictwem wniosku FDA FOIA” …gdzie by się z nimi spotkali następujący mocno zredagowany dokument, gdzie wszystko, co ma jakiekolwiek znaczenie, jest zakryte (b)(4) redakcjami.
Czytanie między wierszami: jest oczywiste, że zarówno producenci, jak i amerykańska FDA nie chcą, aby ktokolwiek poza nimi samymmi znał pełne składniki zastrzyków mRNA lub nawet testował ich czystość i konsystencję.
Według urzędników FDA: Produkcja farmaceutyczna jest Wysoko Skłonność do błędu:
Wiele w procesie produkcji farmaceutycznej coś może – i zdarza się – pójść nie tak. Poza potencjalnymi niespójnościami w przypadku zastrzyków mRNA/LNP, istotne są kwestie jakościowe i ilościowe każdy Produkt farmaceutyczny podlegający regulacjom FDA. Nawet Izba i Senat formalnie potwierdziły doniesienia o niepowodzeniu FDA w zabezpieczeniu amerykańskiego łańcucha dostaw farmaceutycznych. Wiekszosc z Farmacja amerykańska produkt konsumenta końcowegojest produkowany za granicą w krajach takich jak Indie i Chinyi inne kraje o niskich kosztach pracy nie ceniony za wysoki poziom kontroli jakości. Rejestr Federalny jest pełen raportów dot naruszeń w indyjskich i chińskich zakładach produkcyjnych.
Czy FDA certyfikuje również te zakłady – w tym te, które mają długą historię naruszeń – za pośrednictwem systemu przesyłania pocztą do FDA? Skandaliczne jest, że odpowiedź na to pytanie sprawi, że każdy, kto interesuje się jakością farmaceutyczną, poczuje się bardzo niekomfortowo.
Podczas Six Sigma poziom precyzji od dawna był celem w zakresie jakości i bezpieczeństwa w samochodach, komputerach, telefonach komórkowych i innych zaawansowanych technologicznie produkcjach, wydaje się, że był on najczęściej pomijany w przypadku produkcji farmaceutycznej.
Urzędnicy FDA opublikowali dane szacujące niedokładność w produkcji farmaceutycznej na poziomie 2–3σ (sigma). Odpowiada to jakości 2σ 308,537 1,000,000 defektów na XNUMX XNUMX XNUMX możliwości. (Prawdopodobnie istnieje znacznie więcej niż 1,000,000 XNUMX XNUMX możliwości popełnienia błędu w produkcji farmaceutycznej). FDA jest tego świadoma na najwyższych szczeblach kierownictwa; właściwie prąd Szef Urzędu Jakości Farmaceutycznej FDA, Michael Kopcha nawet napisał i opublikował powyższe obliczenia Six Sigma, ubolewając nad nieprecyzyjnym charakterem produkcji farmaceutycznej z powrotem w 2017.
Szerokość błędu dla produktów mRNA i/lub ich LNP może być równa mniej dokładne niż 2-3σ (im niższe σ, tym bardziej błędny jest produkt), ponieważ zawierają materiał nukleotydowy i nowe LNP, co czyni je znacznie bardziej złożonymi niż małocząsteczkowe farmaceutyki – pomimo ich opracowywania, wytwarzania i wypuszczania na rynek „ prędkość warp."
Nawet jeśli FDA i jej urzędnicy uznali nieodłączną niedokładność produkcji, dlaczego w szerokim świecie sportu czy FDA nie wypełnia swojej misji w zakresie bezpieczeństwa, udostępniając publicznie wyniki testów uwalniania technologii mRNA amerykańskiemu społeczeństwu, które je finansuje?
Znowu przed 1862 rokiem? Czy zastrzyki mRNA to jedyne narkotyki, na które Amerykanie nie mają? Absolutna Informacje o składnikach?
Brak jasności co do liczby sekwencji zastrzyków mRNA i innych krytycznych informacji stanowi bezpośredni kontrast w przypadku innego leku na bazie RNA zatwierdzonego przez FDA – patisyran (Onpattro®). Onpattro w przejrzysty sposób podaje sekwencję, masę cząsteczkową i moc w miligramach swoich produktów w ramach oficjalnej FDA oznakowanie opakowania jak pokazano w poniższym fragmencie:


Brak swoistości dawki mRNA Covida: 0.3 ml (lub 0.5 ml) czego?
W tej chwili nadal nie mamy podstawowych informacji o składnikach jakiegokolwiek zastrzyku mRNA Covida. Farmaceuci wiedzą tylko, żeby podać konkretny Tom płynu i najwyraźniej zrobił to bez żadnych pytań. Zwykle oficjalne etykiety opakowań FDA powinny zawierać szczegółowe informacje na temat rzeczywistych składników w danej objętości, ale nie w przypadku etykiet mRNA Covida: po prostu podają 0.3 ml (lub 0.5 ml) jako „postać i moc dawkowania”.
Ponadto, jak może powiedzieć każdy uczeń szkoły średniej, 0.3/0.5 ml to ok Tom, nie a jest determinacja.. Nie znamy żadnych szczegółów ilościowych dotyczących zawartości tego 0.3/0.5 ml, np.: Ile cząstek LNP? Jaki rozmiar/morfologia tych LNP? Ile sekwencji mRNA znajduje się w tej objętości?




Czy to jest uznawane przez FDA za wystarczająco przejrzyste lub „prawdziwe oznakowanie”?
Powyższy fragment „kopiuj i wklej” z ulotki dołączonej do opakowania zawiera wszystkie informacje, którymi producenci dzielą się z konsumentami dotyczącymi dawki – która jest żałośnie niewystarczająca w porównaniu ze wszystkimi innymi etykietami FDA – lub z każdym, kto chce dowiedzieć się czegokolwiek poza ilością płynu. do wstrzyknięcia oraz stężenie 30 lub 100 mcg nieokreślonej sekwencji mRNA.
Niezwykła niedokładność tej etykiety, dozwolona przez FDA, wydaje się być sprzeczna z jej prawie 120-letnią etykietą, w szczególności: „wymaganie, aby żywność i leki były opatrzone zgodnymi z prawdą oświadczeniami na etykietach i spełniały określone standardy czystości i wytrzymałości".
Czy to jest to, co FDA uważa za „prawdziwą” listę składników? (Widzieć 21CFR §352, 21 CFR §201.10 dotyczące „oświadczenia o składnikach” oraz „leków i wyrobów błędnie oznakowanych”).
Pytanie brzmi: czy wymienianie nieznanych lub niespecyficznych składników jest takie, że nikt poza producentem nie jest w stanie ich rozszyfrować naprawdę spełniają ducha lub wymogi prawne dotyczące „etykietowania”? Czy amerykańska FDA uważa tę etykietę za „prawdziwą”? Tak czy inaczej po czyjej stronie stoi FDA; producenci czy konsumenci?
Oprócz tego, że nie jest to bezpośrednio określone, dokładnej liczby nici LNP ani mRNA w zastrzyku 30 lub 100 mcg nie można nawet ekstrapolować stechiometrycznie lub na podstawie Numer Avogadro, ponieważ sekwencja mRNA, masa cząsteczkowa i/lub składnik/konfiguracje LNP nie są nigdzie podane na oficjalnym oznakowaniu FDA.
Skąd ktoś może wiedzieć, czy liczba nici mRNA kodujących białko kolczaste dla Covida jest proporcjonalna do ilości inokulum Covid, które można otrzymać w wyniku infekcji nabytej poza społeczeństwem? Odpowiedź: oni nie mogą.
Czy zastrzyki mRNA Covida Odpowiednio oznakowane/błędnie oznakowane?
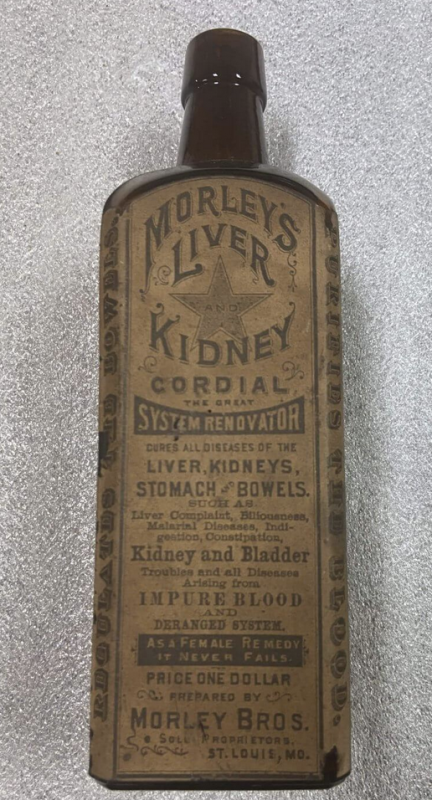
21 CFR 211.125 określa „Należy sprawować ścisłą kontrolę nad etykietami wydawanymi do użytku w operacjach etykietowania produktów leczniczych,”, ale wydaje się, że FDA była tak pobłażliwa, jeśli chodzi o zatwierdzone etykietowanie zastrzyków mRNA Covida, pomimo faktu, że każdy inny lek – w tym Onpattro na bazie mRNA – podaje te informacje. Historycznie rzecz biorąc, decyzje regulacyjne FDA (takie jak informacje, które należy uwzględnić na etykiecie produktu) opierają się na pierwszeństwie, a szczepionki mRNA Covida stanowiły oczywiste odchylenie od historycznego i prawnego pierwszeństwa FDA. Ten godny uwagi brak danych i niejasność nawiązują do czasów: Kordiał Morleya na wątrobę i nerki pod koniec XIX wieku. Różnica polega na tym, że wtedy nie istniała FDA, ale dziś istnieje FDA zatrudniająca około 1800 20,000 pracowników, z których przynajmniej niektórzy rzekomo wierzyli, że ta etykieta jest przejrzysta i „prawdziwa”.
Podanie nieznanego/nieczytelnego/niejasnego składnika, którego nikt nigdy nie był w stanie dokładnie określić, prawdopodobnie nie jest tym, co mieli na myśli prawodawcy z 1906 r. Ustawa o czystej żywności i lekach, określając zasady FDA dotyczące „prawdziwego etykietowania”. Poza tym: fakt, że dawki są podwojone na objętość od różnych producentów (30 mcg/0.3 ml vs 100 mcg/0.5 ml) oznacza, że te sekwencje mRNA wydają się znacznie różnić pod względem długości nukleotydów, co z kolei musiałoby coraz więcej różnych LNP i załączników. Czy to oznacza, że sekwencje mRNA użyte do transkrypcji białka kolczastego są około dwukrotnie większe (10 mcg/0.1 ml w porównaniu z 20 mcg/0.1 ml) w porównaniu z sekwencjami różnych producentów, czy też coś innego przyczynia się do różnicy w długości nukleotydów?
Dla laika, który przeczytał do tego momentu (swoją drogą, brawa): brak szczegółowych informacji na etykiecie mógłby przypominać szerszą reklamę domu na sprzedaż, stwierdzającą, że jest on zbudowany z drewna i cegieł, na płycie cementowej – ale nie pokazującą żadnych zdjęć domu (np. sekwencji) i nie udostępniania jego powierzchni (np. masy cząsteczkowej). W każdym razie brak informacji jest niewystarczający i stanowi odstępstwo od tradycyjnych standardów.
Każdy inny lek zatwierdzony przez FDA – w tym inne leki mRNA – zawiera pełne ujawnienie składników swoich produktów, w tym reprezentacja strukturalna i masa cząsteczkowa produktu, aby ludzie dokładnie wiedzieli, co dostają.
To prawda: poszukaj w wyszukiwarce dowolnego narkotyku, jaki przyjdzie Ci na myśl Baza danych Drugs.com i zwróć uwagę, jak wszystkie znaczniki zapewniają strukturę i/lub masę cząsteczkową. Dowód na to, że zastrzyki mRNA Covida są: wyraźny wyjątek od historycznej praktyki zatwierdzania przez FDA i zasady „prawdziwej etykiety”.
Szczegóły badania duńskiego 2023 Znacząca zmienność kliniczna pomiędzy seriami zastrzyków mRNA Covid-19 mRNA:
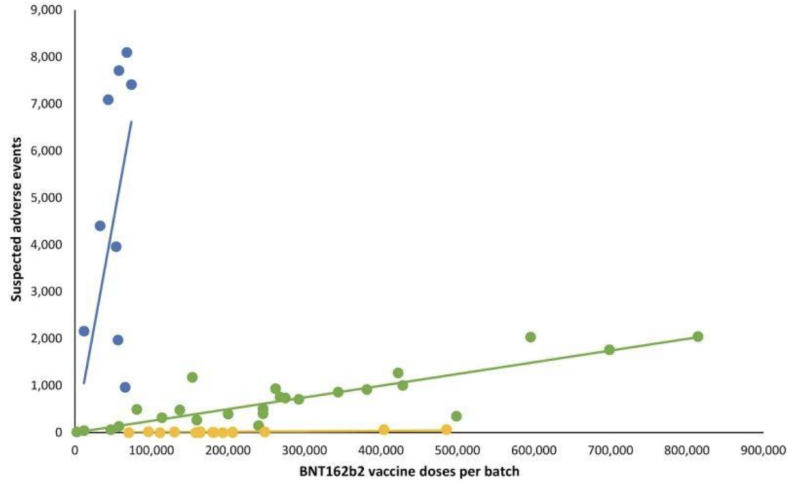
Wydaje się, że brak przejrzystości nawet w przypadku potencjalnie nieprawidłowej walidacji testów „przesłanych pocztą” dał producentom przepustkę do kolejnej niezwykle ważnej części tego, co nadzoruje FDA: potencjalnych objawów klinicznych dotyczących różnic w partii zastrzyków mRNA. Retrospektywa Duńskie badanie bezpieczeństwa opublikowany wcześniej w 2023 r. szczegółowo opisywał wysoce odbiegający wzór raportów o zdarzeniach niepożądanych po wstrzyknięciach mRNA BNT162b2 firmy Pfizer-BioNTech, skorelowany z duńskim systemem zgłaszania zdarzeń niepożądanych DKMA.
Na poniższym wykresie liniowym różne kolorowe kropki reprezentują różne partie zastrzyków mRNA firmy Pfizer-BioNTech. Podzielił partie na trzy różne kategorie; wysoka-niska- do (prawie) nieobecna liczba zgłoszonych grup zdarzeń niepożądanych (odpowiednio niebieski, zielony i żółty wykres).
Innymi słowy: wydaje się, że przypuszczalnie „równoważne” produkty tego samego producenta charakteryzują się bardzo różną częstością występowania zdarzeń niepożądanych w zależności od partii, przy czym każda z tych partii reprezentuje setki tysięcy wstrzyknięć mRNA.
Po dodaniu odpowiednich linii regresji liniowej wyłonił się konkretny wzór:
Ważne pytania dotyczące godnych uwagi różnic w zdarzeniach niepożądanych pomiędzy partiami mRNA Covid-19 obejmują:
- Czy różnice w zdarzeniach niepożądanych mogą wynikać z jakościowych lub ilościowych różnic w sekwencjach mRNA lub liczbie nici mRNA pomiędzy partiami?
- Czy różnice w zdarzeniach niepożądanych mogą wynikać z jakościowych lub ilościowych różnic w wielkości/morfologii lub ilości LNP pomiędzy partiami? Jakie badania zostały wykonane zapewnić bezpieczeństwo różnych LNP stosowane w zastrzykach mRNA?
- Czy te partie, które odpowiadały punktom danych żółtym, zielonym i niebieskim, różniły się w jakiś sposób jakościowo lub ilościowo?
- Czy przechowywanie/obsługa poprodukcyjna w zakładzie administrującym (lub gdziekolwiek indziej w łańcuchu dostaw) była zagrożona, co doprowadziło do zmienności produktu?
- Jaka jest Sigma/stopień błędu tego i innych produktów pochodzących z konkretnego zakładu produkcyjnego/szefa zmiany odpowiedzialnego za produkcję?
- Czy składniki produktów mRNA Covida pochodziły z Indii lub Chin, a nie z innych źródeł, w zależności od partii?
- Jaki procent partii produktów mRNA Covid został przetestowany poprzez odbiór osobisty przez inspektora FDA w porównaniu z partiami „wysłanymi” od początku do chwili obecnej? Czy każdą pojedynczą partię badano przy użyciu tylko jednej z tych dwóch metod zbierania?
- Czy FDA przeprowadziła weryfikację testów dopuszczenia do obrotu partii duńskiego systemu zgłaszania zdarzeń niepożądanych DKMA? Jeśli tak, dlaczego FDA nie publikuje wyników tych konkretnych testów? Jeśli nie, dlaczego nie przeprowadzono testów?
- Czy istnieje zasadniczy problem w konsekwentnym wytwarzaniu sekwencji LNP i/lub mRNA w sposób niezawodny i pozbawiony zanieczyszczeń?
Wyniki duńskiego badania i powyższe pytania dotyczące zdarzeń niepożądanych można *zacząć* rozpatrywać, ale nie bez niezależnego udostępnienia przez FDA wyników swoich wyników testów uwalniania. W obecnej sytuacji, ze względu na wszechobecne redakcje FDA (b) (4), nikt nie zna zatwierdzonej metodologii testowania zastrzyków mRNA Covida or które dokładnie partie w duńskim badaniu zostały przetestowane, a które nie or wyniki tych testów partii.
…Z drugiej strony, nawet gdyby FDA zdecydowała się opublikować wyniki testów serii, skąd konsumenci mają wiedzieć, czy wyniki te są reprezentatywne dla określonych serii, skoro producenci sami wybierają, które próbki mają zostać „wysłane pocztą”?
Niezapewnienie przejrzystości składników i zapewnienie jakości poprzez odpowiednią metodologię pobierania próbek jest podstawowym i podstawowym wymaganiem FDA. W rzeczywistości był to główny powód powstania FDA! Czy Amerykanie nie zasługują na lepszą przejrzystość, nadzór i przepisy dotyczące „prawdziwego etykietowania” naszych produktów farmaceutycznych – zwłaszcza, że te przepisy wprowadzono ponad 100 lat temu?
Opublikowane pod a Creative Commons Uznanie autorstwa 4.0 Licencja międzynarodowa
W przypadku przedruków ustaw link kanoniczny z powrotem na oryginał Instytut Brownstone Artykuł i autor.








